
Vibrational and Electronic Spectra
The Electronic Ground State
Molecules are almost always in their electronic ground state, that is, the electrons fill the orbitals of the molecule according to the Aufbau Principle, orbitals of lower energy are filled before orbitals of higher energy. If the orbitals are not filled in this manner, the molecule is in an electronic excited state. This is depicted below for the hydrogen atom (Figure 1).
A molecule in the electronic ground state can exist in a variety of vibrational and rotational states. The ground state with all of these vibrational and rotational states is represented by a Morse-like curve shown in Figure 2. The x-axis represents the so-called Q coordinate and the y-axis represents the energy of the molecule. For the hydrogen atom, Q can be thought of as the internuclear distance between the two hydrogen atoms. For very small values of Q, the energy is very high due to repulsion of the positively charged nuclei. Very large values of Q result in isolated, noninteracting atoms and the energy represents the energy of the isolated atoms. In between the extremes is the region of bonding interactions between the atoms.

Vibrational Excited States
As mentioned above, a molecule in the ground electronic state can exist in a variety of vibrational and rotational states. In this tutorial, only vibrational states will be covered. Each vibrational state exists at a definite energy, but over a range of Q values. Existing at a definite energy and definite Q-value would violate the Heisenburg Uncertainty Principle because both the location of the atoms and their energy could be known precisely at the same time. For this reason, the lowest vibrational state is not at the absolute bottom of the electronic "well" as shown in Figure 3. Transitions between vibrational levels can oocur upon absorption of a photon as shown in Figure 4. The photon can change the vibrational level (v) by 0, +1, or -1. You may be wondering what this has to do with the inorganic lab. The energy difference between the vibrational levels falls in the infrared region of the electromagnetic spectrum. The absorptions observed in the infrared spectrum are changes in these vibrational levels within one electronic state.


Sometimes these vibrational absorptions are very localized and can be associated with the stretching or bending of a specific bond. Most of the time,however, the individual bond stretches and bends are coupled. The observed IR absorptions are combinations of the bending and streching of several bonds. Symmetry and relative energies determine the combinations that are observed, but in a relatively complicated matter (Group Theory books cover this area). In general, the more symmetry equivalent bonds to vibrate there are, the fewer the absorptions that will be observed. There may still be more than one absorption observed for a group of symmetry equivalent bond vibrations.
Carbon Monoxide Ligands and Backbonding
One type of bond streching mode deserves special attention here, that of the carbon monoxide ligand (or carbonyl ligand as is is often called). Carbonyl ligand streching occurs in a well defined energy range 1700 cm-1 to 2200 cm-1. Carbonyl stretching modes are often coupled to other carbonyl streches, but not other types of stretches or bending modes. That is, the observed absorptions are fairly "pure" carbonyl stretching modes. As you might have noticed, these stretches occur over a fairly large energy range. This is because the observed stretching frequency is very dependent on the bonding between the ligand and the metal center.
The carbon monoxide ligand bonds to a metal by donating electron density (from it's nonbonding electron pair) into a metal d-orbital of sigma symmetry and accepting electron density from a filled metal d-orbital of pi symmetry into it's pi* antibonding orbital. The frontier orbitals of carbon monoxide are shown below in Figure 6 as pictoral depictions calculatedwith CAChe. As you can see, the HOMO of CO is primarily a lone pair orbital. It is also slightly antibonding between the carbon and oxygen atom. The LUMO on the other hand is a pi*
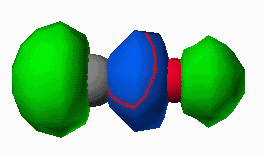
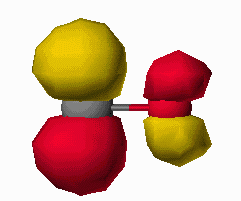
Figure 6. HOMO and LUMO of CO as calculated with CAChe. Gray atom is C, Red atom is O.
orbital (antibonding between the carbon and oxygen atoms. When the CO molecule bonds to a metal, these frontier orbitals interact as shown in Figure 7. The donation of electron density from the CO to the metal results in a slight strengthening of the CO bond (electron density is being removed from a slightly antibonding orbital). Likewise the electron density being accepted into the pi* orbital results in a dramatic weakening of the CO bond. The frequency of a bond vibration in a diatomic molecule is determined by Hooke's Law shown in equation 1. The frequency is proportional to the square root of the force constant of the bond.
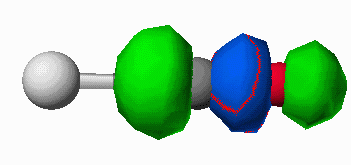 (a)
(a)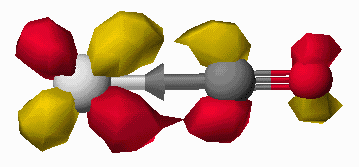 (b)
(b)
Figure 7. Interaction of the CO HOMO (a) and the CO LUMO (b) with a metal atom (silver atom,Mo).

Therefore, the stronger the bond, the higher the energy of the stretching frequency for that bond (for the same molecule). The importance of this, is that the amount of pi donation to the carbonyl ligand is usually dependent on the amount of electron density at the metal center. Therefore, the carbonyl stretching frequencies can be used as an indicator of the electron density of the metal in a closely related set of molecules.
Electronic Excited States
Transitions from one electronic state to another can also occur upon absorption of a photon. The energy range for these transitions falls in the ultraviolet and visible range of the electromagnetic spectrum for the most part. A transition from one electronic state to another is shown in Figure 8. Much information can be drawn from such a figure.

Figure 8. A transition from the ground electronic state to an excited electronic state
Organic Molecules and Selection Rules
You have probably had some background in UV-Vis spectroscopy in your organic class. The types of transitions important to organic molecules are pi-pi*, sigma-sigma* and n-pi*. These transitions are fairly intense because of the selection rules. For molecules with an inversion center, the symmetry selection rule is that the orbital the electron is promoted from and the orbital the electron is promoted to cannot have the same symmetry with respect to the inverision center. To begin with, a molecule has an inversion center if every point in the molecule (x,y,z) can be interchanged with every point (-x, -y, -z) when the center of the molecule is at point (0,0,0) without any noticable change to the molecule. If we take ethylene as an example (Figure 9), the pi orbital has ungerade (u) symmetry with respect to the inversion center. This is because if we transfer every point (x,y,z) to the point (-x,-y,-z), the resulting orbital has the positive lobes replaced by the negative lobes and vice-versa. Similarly, the pi* orbital has gerade (g) symmetry because a n inversion operation on this orbital results in no change. Since, the pi orbital is u and the pi* orbital is g, the pi-pi* transition is symmetry allowed. In the same way, a transition from the C-C sigma bond (g symmetry) to the C-C sigma* orbital (u symmetry) is allowed, but the transition from sigma (g symmetry) to pi* (g symmetry) is symmetry forbidden.
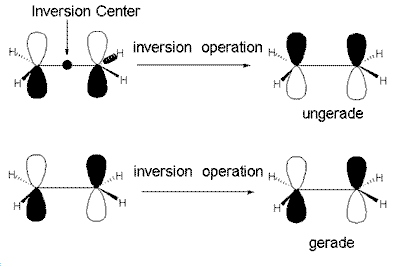
Figure 9. Examples of ungerade and gerade orbitals of ethylene
Another way of looking at these transitions is with molecular orbital diagrams. In figure 10, part of the molecular orbital diagram of ethylene is shown (the C-C sigma and pi bond portions). The same transition that can be shown with the energy well diagram can be shown as excitation of an electron from one orbital to another.

Inorganic Molecules
When there is a metal present in the molecule, several other types of transitions are possible. This is because of the more complex orbital schemes in metal complexes. This first (and most intense) type of metal-based transitions are the charge transfer transitions. In the example of ethylene above, the initial and final orbitals involved in the transition were centered on the same atoms. In a charge transfer transition, they are not (hence charge transfer transitions, the transfer of an electron from one part of the molecule to another). The two types of charge transfer transitions we will discuss are metal to ligand charge transfer transitions (MLCT) and ligand to metal charge transfer transitions (LMCT). In a typical MLCT transition, an electron from one of the metal orbitals is transferred to a pi* orbital of one of the ligands on the metal. If the metal has unoccupied d orbitals, a transfer from a ligand orbital to the metal is also possible (LMCT).
The second type of transition we will discuss are d-d transitions. This is the excitation of an electron from one metal d-orbital to another metal d-orbital. You might have noticed a problem with this type of transition. All of the metal d-orbitals are of g symmetry. That makes a d-d transiton symmetry forbidden. This brings us to vibronic coupling. Certain vibrations can remove the center of symmetry from the molecule. This makes the d-d transitions weakly allowed and in some cases, observable.
Intensity
The d-d transitions from the previous section are formally symmetry-forbidden. This forbidden character manifests itself in the intensity of the observed d-d transitions. Most pi-pi* and sigma-sigma* transitons have intensities in the several hundred to a few thousand L/mole cm. Most d-d transitions have intensities of less than 100 L/mole cm. Charge transfer bands can have very large intensities (in the 10's of thousands).
| Type of Transition | Approximate Intensity (L/mole cm) |
| pi-pi* | 1000 's |
| sigma-sigma* | 1000 's |
| d-d | 10-100 |
| charge transfer | 1000-10000 's |
Reporting Data
For electronic transitions, the information that needs to be reported is the solvent (the transitions are solvent dependent), the wavelength of the maximum absorbtion (epsilon max, usually in nanometers) and the extinction coefficient. The extinction coefficient is not always reported for known compounds. For vibrational transitions, the solvent ,(or matrix) the minimum transmittance of the absorbtion (usually in wavenumbers), and a relative measure of the intensity of the band (vw for very weak, w for weak, m for medium, s for strong, vs for very strong, b for broad). The measures of intensity are guesses and are relative to other peaks of the compound in the spectrum.
Return to the main tutorials page